
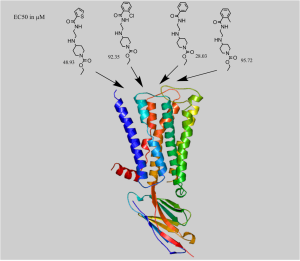
Figure: Docking of a series of allosteric modulators to human Muscarinic receptor type 2 (4MQT)
The discovery of small molecule ligands for protein binding (e.g. drugs, probes) is often split into two questions: “what will bind?” and “how will it bind?”. The “what” problem identifies small molecules that have substantial binding affinity against a given protein structure. Computationally, this is done using a virtual screen that examines molecular databases for compounds chemically similar to known binders. These ligand-based techniques take advantage of experimentally derived structure-activity relationships (SAR). SARs are generated from a series of compounds sharing a common scaffold, all of which have been tested for activity on a particular target. The “how” question looks at important interactions between a protein and its ligand and can be computationally explored through ligand docking. RosettaLigand is an established program for generating models of protein-ligand complexes that consider both protein and ligand flexibility. However, there is often a gap between the virtual screening methods in cheminformatics and the ligand docking methods in structural biology. The new algorithm, RosettaLigandEnsemble, bridges that gap by incorporating information from the “what” question to better answer the “how” question.
RosettaLigandEnsemble will simultaneously optimize the binding mode for a congeneric series of ligands. It utilizes a correlation coefficient to favor sampling of possible binding positions where the Rosetta scoring agrees with experimental SAR data. This takes advantage of the fact that structurally similar active compounds should adopt a similar pose in a binding pocket. Furthermore, the common and unique interactions in said pocket should explain differences in experimental affinity. The ever increasing amount of binding data for compound series presents a timely opportunity for RosettaLigandEnsemble. We hope to apply the extra information in RosettaLigandEnsemble to dock difficult systems, particularly those involving protein homology models.
Alumni Project Members: Darwin Fu