
Uncommon structural features and the absence of any initial guess make structure elucidation of natural products challenging, particularly if crystallization is not an option. Frequently, multiple structural alternatives can be suggested from one- and two-dimensional NMR spectra, but a unique identification of the correct solution is difficult. This calls for an automated method to exhaustively search the chemical space and provide a ranked list of all possible structural solutions for further consideration by an expert chemist.
Focus of this project is to develop a computational tool for automated elucidation of constitution, configuration, and conformation of natural compounds from their NMR data.
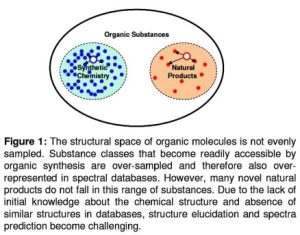
Prediction of spectral parameters such as chemical shift (CS) is one approach to discriminate incorrect structural models. Most of the current CS prediction methods rely on large spectral and structural databases. These databases sample the natural product space sparsely leading to increased inaccuracy in CS prediction (Figure 1).
In addition some of these databases are not readily available to academia. An artificial neural network (ANN) is proposed to rapidly and accurately predict the 13C, 1H, and 15N chemical shifts from the structure of small organic molecules. In the sparsely sampled space of natural compounds, the excellent interpolation abilities of ANNs will improve the prediction accuracy.
Figure 1
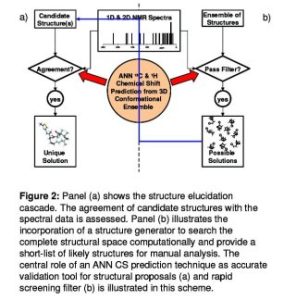
To deal with the stereochemically challenging structure of most natural products, I want to include spatial information of organic compounds (Figure 2) . E.g., the probability of certain dihedral angles will be described by energy functions constructed from histograms of substances with known 3D structures. The Cambridge Structure Database (CSD), the largest for 3D X-ray structures of organic compounds, is used to obtain this 3D information.
Figure 2
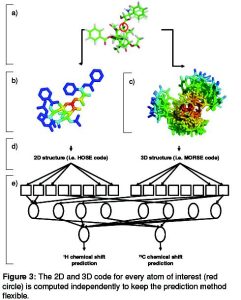
The training data for the ANN is extracted from the NMRshiftDB, an open database for 13C and 1H spectra of organic compounds. The inclusion of the proton CS will allow me to use information from multi-dimensional NMR to further increase the accuracy. Furthermore I want to give the possibility to include known functional groups into the description of the unknown compound. Therefore a list of common structural features (fragments) was developed.
Figure 3
Alumni Project Members: Ralf Mueller, Laura Wiley, Nils Woetzel