
It is the objective of the present project to map structure-activity relation (SAR) data into a 3D pharmacophore map using a novel computational algorithm “PHARMMAP”. The novel architecture of PHARMMAP utilizing a Monte Carlo random walk structure generator and QSAR models based on machine learning techniques will allow the subsequent design of small molecules focused libraries with improved activities for chemical synthesis.
The project aims to both a) build innovative ChemInformatics software which can be adopted within the NIH Molecular Libraries programs, and b) elucidate novel allosteric potentiators of mGluR5 for the potential treatment of schizophrenia. In contrast to other pharmacophore mapping approaches2-5 the PHARMMAP algorithm requires no crystal structure of the target protein. Therefore, this method can be applied to membrane proteins which are the target of 40-50% of modern therapeutics but have often no high-resolution crystal structure available.
Recent data suggest that activation of metabotropic glutamate receptor subtype mGluR5, which is responsible for the regulation of neuronal circuits involved in disorders that disrupt cognitive function, may prove useful in treating such disorders. The latest generation of selective mGluR5 potentiators features systemically active compounds able to cross the blood-brain barrier. Further development of selective potentiators of mGluR5 may provide an exciting and novel avenue for the treatment of schizophrenia.
A high-throughput screen (HTS) for mGluR5 potentiators at Vanderbilt’s molecular libraries screening center network facility revealed ~1,400 diverse compounds in a library of ~150,000 whose activity was validated by independent experiments. It is my hypothesis that the relation of chemical structure and biological activity reflected in these screening results can substantially contribute towards understanding the structural basis for mGluR5 potentiation.
Aim I: Develop PHARMMAP Machine Learning QSAR Models and a Monte Carlo Random Walk Structure Generator
- Train QSAR models on HTS data using ANN and SVM algorithms
- Implement Monte Carlo random walk structure generator for pharmacophore mapping and derivative design
- Benchmark novel approach against fragment-dependent Comparative Molecular Field Analysis
Aim II: Drive Iterative Cycle of Hit-to-Lead Optimization
- Collaborative hit selection, design of focused library of hit derivatives, and synthesis
- Refine QSAR models and pharmacophore map to enable iterative hit-to-lead optimization
- Dissemination through WWW based server interface and free licensing of software for academic use
The research strategy bases on the development of cheminformatics methods that tackle critical limitations of current tools such as accurate but rapid, scaffold-independent pharmacophore mapping. However, going beyond a pure method development project, the present research strategy intimately links the developed methods to application in drug discovery and also – to a smaller extent – to investigating biology and pharmacology of mGluR5.
This project links drug discovery with biological research as we apply the cheminformatics tools in both settings. We see this as an advantage as biological research will inform drug discovery and vice versa – for example structural determinants of the mGluR5 allosteric sites are likely to inform the hit-to-lead optimization process and compounds designed in the hit-to-lead optimization process can become useful probes of a biological system. The overall project outline is illustrated in Figure 1.
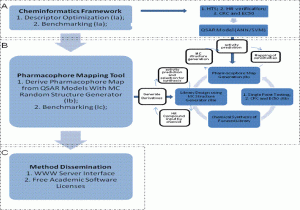
Figure 1: Overall research strategy where cheminformatics method development informs drug discovery. Depicted are the use of a cheminformatics framework for the identification of hit compounds with novel chemotypes, and the implementation of a pharmacophore mapping tool for the design of focused libraries during hit-to-lead optimization of mGluR5 PAM. (A) Cheminformatics framework for vHTS using QSAR models implemented in the mentor’s lab1. (B) Implementation of a pharmacophore mapping tool and its application in the design of focused libraries during hit-to-lead optimization of mGluR5 PAMs. (C) Method Dissemination strategy.
Alumni Project Members: Edward W. Lowe Jr, Alexander Geanes