
Virtual screening is a computational technique that aids the drug discovery process. The focus of this project is ligand-based screening where, in contrast to structure based screening, knowledge about the biological target structure is not necessary. This way hits of novel chemical structures can be determined that bind to a macromolecular target of interest, like receptors or enzymes.
Large compound libraries were tested in High Through-put screens for biological activity and serve as a knowledge base. Chemical structures were encoded by molecular descriptors and used as input to machine learning techniques like Artificial Neural Networks (ANNs), Support Vector Machines (SVMs) to develop quantitative structure–activity relationship (QSAR) models.
QSAR is an area of computational research that builds virtual models to seek quantitatively correlate complex non-linear relations between chemical and physical properties of a molecule with its biological response, such as activity for a specific biological target.
This technology allows rapid screening in large compound libraries of small organic molecules to find novel structures that are most likely to bind a desired drug target in silico.
The aim of this project is the method development of ChemInformatics tools for QSAR modeling in the BCL.
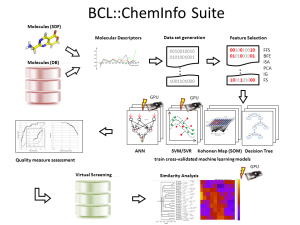
Figure 1: BCL::Cheminfo: A virtual high-throughput screening suite that provides a workflow from molecule preparation, data set generation, QSAR model training, cross-validation, and analysis to a virtual screening of large compound libraries.
In exciting recent developments, we have applied the techniques of Deep Learning to QSAR and found >30% improvement in enrichments using these methods versus conventional techniques.
QSAR models require descriptors that accurately convey chemically-relevant information to the machine learning models. We recently enhanced 2D and 3D autocorrelations by incorporating sign-sensitivity into them, in particular for charge-based descriptors. This addition leads to an additional 15% relative improvement in logAUC across a large benchmark set (2015. Sliwoski, Mendenhall, Meiler. J Comp Aided Mol Design).

SMILES strings for molecules used in the manuscript 2016 Mendenhall JL, Meiler J; “Improving Quantitative Structure-Activity Relationship Models using Artificial Neural Networks Trained with Dropout.” J Com Aid Mol Design as well as 2015 Sliwoski G, Mendenhall J, Meiler J “Autocorrelation descriptor improvements for QSAR: 2DA_Sign and 3DA_Sign” J Com Aid Mol Design. Molecules are labeled with their PubChem compound ID, wherever available. All molecules have been subject to the standardization procedures described in the manuscripts.
Current Project Members: Oanh Vu
Alumni Project Members: Mariusz Butkiewicz, Ralf Mueller, Eric Dawson, Edward W. Lowe Jr, Alexander Geanes, Jeff Mendenhall, Gregory Sliwoski