
Learning systems have a long history in being applied to reduce the search space for protein structure prediction e.g. artificial neural networks (ANN) and hidden markov models (HMM) are used for protein secondary structure prediction and motif recognition. Recently, ANNs were successfully utilized to derive a consensus amino acid contact prediction for unknown folds from fold recognition techniques. These predictions drive de novo prediction of protein tertiary structure towards better results by reducing the conformational space. A highlight of this work is the coupled prediction of protein secondary and tertiary structure, where formation of secondary structure and tertiary structure drive each other.
We plan to built on our experience in developing such methods when taking these methods to the next level. While above methods predict backbone structure at low-moderate resolution, we plan to develop a high resolution ANN contact prediction, from protein sequence only. This method will predict direct amino acid side chain interactions between mutually packed secondary structure elements (SSE), which can be used later to improve high resolution protein structure prediction.
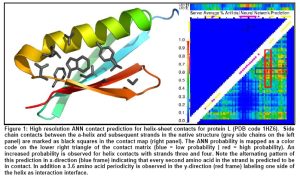
Rational behind attempting a prediction of such secondary structure element (SSE) contacts is the knowledge about the intimate packing of side chains between these SSEs which define and stabilize the protein fold. In a β-strand every other side chain will point towards the core of such an interaction and can participate in packing. Within an α-helix stretches of 1-3 amino acids per winding have the opportunity to interact within one interface. The helix periodicity of 3.6 amino acids will be reflected in these interaction patterns. Most prominent among such interaction motives is the leucine zipper – a recurring sequence pattern with Leucine in every 7th position – which is frequently observed in helix-helix interfaces. This and similar interaction motifs should be detectable from two stretches of amino acid sequence, since the well-defined interaction patterns require specific amino acid sequences in order to be possible.
The ANN requires an input of two sequence windows spanning the potentially interacting SSEs, having the two directly contacting amino acids in the center. In a preliminary experiment, the length of these sequence windows was chosen to be 9 residues for α-helices and 5 residues for β-strands. In result both SSEs have about the same length of 12Å for the interaction interface. For each amino acid in these windows predicted secondary structure, position specific scoring matrices from PSI-BLAST, and a property profile are used as input. Five separate ANNs were trained for helix-helix, helix-sheet, sheet-helix, sheet-sheet, and strand-strand interactions. In result these networks can specialize on the high resolution characteristics of these interactions. A non-redundant fold database (less than 25% sequence identity) is used for training, consisting of ~1800 proteins. All amino acid pairs within one fold that are in contact are used for training. The dataset is balanced with an equal number of non-contacts. An example prediction from a preliminary test network is shown in Figure 1.
Expected outcome.
It is expected that the high-resolution training of the ANNs will amount in a more accurate prediction of contacts, which will in turn result in a drastic reduction of the search space for de novo structure prediction. This reduction will not only affect the low-moderate resolution search of the backbone conformational space. It will also affect high resolution side chain building and structure refinement, since in contrast to earlier approaches specific contacts between amino acid side chains are predicted (instead of larger regions that are likely to be in contact). The output of the ANN can be translated into a potential and used as additional component in the scoring function fro de novo protein structure prediction. Use of contact predictions to reduce the search space for backbone and side chain conformations at the same time, takes contact prediction to the next level.
Alumni Project Members: Mert Karakas, Nils Woetzel, Marcin J. Skwark