
Membrane transport is essential to many areas of cellular life. The movement of ions across the cell membrane is carried out by ion channels which play crucial roles in many physiological processes such as electrical signaling in the brain, muscular contraction, generation of the heartbeat, and hormone secretion.
Voltage-gated potassium (Kv) channels are tetrameric membrane proteins that selectively conduct K+ ions across the cell membrane. They open and close in response to changes in transmembrane voltage. Of the 40 human Kv channels, the KCNQ1 (Kv7.1) channel is special because of its wide range of physiological behaviors in both excitable cells such as cardiomyocytes and non-excitable cells such as epithelia. Mutations in the KCNQ1 gene are associated with different forms of heart arrhythmia such as long QT syndrome (LQTS), atrial fibrillation, and Romano-Ward syndrome, as well as impairment of intestinal chloride ion secretion related to cystic fibrosis.
Research in the Meiler lab studies the structural and dynamical properties of the KCNQ1 channel, e.g., how it interacts with auxiliary proteins and small-molecule drugs, how it facilitates ion permeation, and how it transitions between open and closed states. Using Rosetta protein-protein docking, we previously developed structural models of KCNQ1 in complex with KCNE1 [1] and KCNE3 [2], which suggested how these proteins modulate KCNQ1 activation. We have also quantitatively predicted energetic perturbations in genetic variants of the KCNQ1 channel [3]. The results support the notion that protein destabilization and trafficking defects are a common cause of KCNQ1 dysfunction in LQTS [4].
To distinguish more accurately disease-causing from benign KCNQ1 variants, we develop machine learning models that predict the likelihood that a mutation in KCNQ1 will lead to channel dysfunction [5]. Variant interpretation tools like this could help to improve the medical decision-making for LQTS.
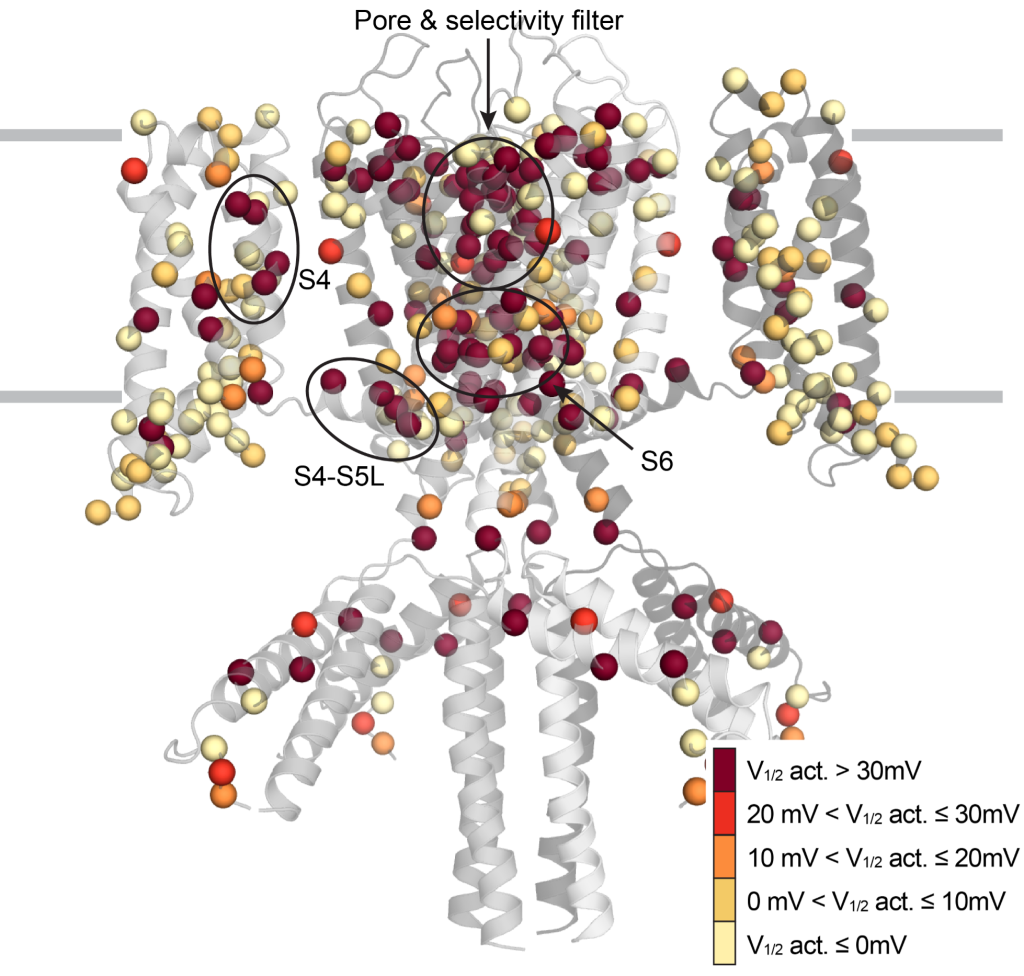
Figure: Location of genetic variants in the KCNQ1 channel with changes in the activation potential.
References:
[1] Kuenze G, Vanoye CG, Desai RR, Adusumilli S, Brewer KR, Woods H, McDonald EF, Sanders CR, George AL Jr, Meiler J. Allosteric mechanism for KCNE1 modulation of KCNQ1 potassium channel activation. Elife. 2020 Oct 23;9:e57680. doi: 10.7554/eLife.57680. PMID: 33095155; PMCID: PMC7584456.
[2] Kroncke BM, Van Horn WD, Smith J, Kang C, Welch RC, Song Y, Nannemann DP, Taylor KC, Sisco NJ, George AL Jr, Meiler J, Vanoye CG, Sanders CR. Structural basis for KCNE3 modulation of potassium recycling in epithelia. Sci Adv. 2016 Sep 9;2(9):e1501228. doi: 10.1126/sciadv.1501228. PMID: 27626070; PMCID: PMC5017827.
[3] Kuenze G, Duran AM, Woods H, Brewer KR, McDonald EF, Vanoye CG, George AL Jr, Sanders CR, Meiler J. Upgraded molecular models of the human KCNQ1 potassium channel. PLoS One. 2019 Sep 13;14(9):e0220415. doi: 10.1371/journal.pone.0220415. PMID: 31518351; PMCID: PMC6743773.
[4] Huang H, Kuenze G, Smith JA, Taylor KC, Duran AM, Hadziselimovic A, Meiler J, Vanoye CG, George AL Jr, Sanders CR. Mechanisms of KCNQ1 channel dysfunction in long QT syndrome involving voltage sensor domain mutations. Sci Adv. 2018 Mar 7;4(3):eaar2631. doi: 10.1126/sciadv.aar2631. PMID: 29532034; PMCID: PMC5842040.
[5] Li B, Mendenhall JL, Kroncke BM, Taylor KC, Huang H, Smith DK, Vanoye CG, Blume JD, George AL Jr, Sanders CR, Meiler J. Predicting the Functional Impact of KCNQ1 Variants of Unknown Significance. Circ Cardiovasc Genet. 2017 Oct;10(5):e001754. doi: 10.1161/CIRCGENETICS.117.001754. PMID: 29021305; PMCID: PMC5679743.